

من در حال حاضر با مشکل زیر گیر کردم:
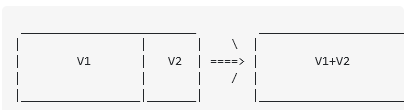
در یک جعبه دو گاز مختلف در دو محفظه وجود دارد که توسط یک دیوار جامد از هم جدا شده اند. دما T
هر دو گاز یکسان است و هر دو گاز از ذرات 1 مول تشکیل شده اند. بنابراین در سمت چپ جعبه، حجم V1 با فشار p1 و در سمت راست حجم V2 و فشار p2 داریم.. نه ما دیوار جداکننده را حذف می کنیم.
اجازه دهید ابتدا فرض کنیم که گازهای مختلفی در سمت چپ و راست داریم. هدف من محاسبه تغییر آنتروپی است که به دلیل برداشتن دیوار رخ می دهد. اولین روش من این بود:
$\Delta S_1 = nR\ln(\frac{V_1+V_2}{V_1})$
$\Delta S_2 = nR\ln(\frac{V_1+V_2}{V_2})$
جایی که ΔSتغییر آنتروپی سیستم، n تعداد مول و R ثابت گاز جهانی تغییر کل آنتروپی اکنون مجموع هر دو خواهد بود:
$\Delta S_{tot} = S_1 + S_2 = nR\ln(\frac{(V_1+V_2)^2}{V_1V_2})$
بعد از مدتی به شکل دیگری به مشکل فکر کردم: ابتدا دیوار را به صورت هم دما حرکت می دهم تا V1=V2
. تغییر آنتروپی ناشی از حرکت به صورت زیر خواهد بود:
$\Delta S_{mov1} = nR\ln(\frac{V_1+V_2}{2V_1})$
$\Delta S_{mov2} = nR\ln(\frac{V_1+V_2}{2V_2})$
$\Delta S_{mov} = S_{mov1} + S_{mov2} = nR\ln(\frac{(V_1+V_2)^2}{4V_1V_2})$
حالا دیوار را جدا می کنم و به دلیل اختلاط گازها تغییر آنتروپی دریافت می کنم:
$\Delta S_{rem} = R(n_1 ln(\frac{n_1+n_2}{n_1}) + n_2 \ln(\frac{n_1+n_2}{n_2})) = 2nR\ln(2)$
بنابراین $\Delta S_{tot} = \Delta S_{rem} + \Delta S_{mov} = nR\ln(\frac{(V_1+V_2)^2}{4V_1V_2}) + 2nR\ln(2)$
که به وضوح با نتیجه ای که من از رویکردم در اولین روش گرفتم متفاوت است. با این حال، از آنجایی که آنتروپی یک متغیر حالت است، نباید این اتفاق بیفتد. نه من گیر کردم من به نتیجه دوم تمایل دارم، اما خیلی دور از اطمینان هستم. خیلی خوب می شود اگر توضیح دهید کدام روشم درست است .
علاوه بر این اگر من گازهای یکسانی در V1 و V2 داشتم
، آنتروپی چگونه رفتار می کند؟ از آنجایی که فشار بعد از برداشتن دیوار تغییر می کند، می گویم آنتروپی نیز تغییر می کند. درست مانند روش دوم من، اما بدون اصطلاح اختلاط. آیا این درست است؟
من تازه متوجه شدم که$2nRln(2)$را می توان به صورت $nRln(4)$ نوشت . از این رو
$nR\ln(\frac{(V_1+V_2)^2}{4V_1V_2}) + 2nR\ln(2) = nR\ln(\frac{(V_1+V_2)^2}{V_1V_2})$
بنابراین هر دو روش من نتیجه یکسانی می دهند. ، سوال دوم من باقی می ماند. آیا در صورت وجود دو گاز یکسان، آنتروپی مطابق با متد دوم من (بدون آنتروپی اختلاط) تغییر می کند؟
آنچه که شما آن را خراب کردید، "Paradox Gibbs" نامیده می شود، و قانون این است که فضای فاز را برای محاسبات آنتروپی در مکانیک آماری با عامل ذرات یکسان تقسیم کنید، که تعداد پیکربندی را کاهش می دهد.
از آنجایی که درجه حرارت در این فرآیند تغییر نکرده است، توزیع حرکت اتم ها بی اهمیت است، همان قبل و بعد از آن است و آنتروپی کاملا فضایی است، همانطور که متوجه شدید. حجم فضای پیکربندی برای قسمت چپ این است:
${V_1^N \over N!}$و برای بخش راست این است:${V_2^N\over N!}$و حجم کل فضای پیکربندی ذرات 2N است:
$(V_1V_2)^N\over (N!)^2$هنگامی که مانع می شود، حجم فضایی فضای پیکربندی را دریافت می کنید$(V_1 + V_2)^{2N} \over (2N)!$هنگامی که v1و v2برابر هستند، شما به طور انحصاری انتظار می رود صفر آنتروپی افزایش یابد. اما شما با از بین بردن دیوار کمی کمی از آنتروپی کمی به دست می آورید. قبل از اینکه دیوار را برداشتید، تعداد ذرات در سمت چپ و سمت راست دقیقا برابر بود، در حال حاضر آنها می توانند کمی نوسان کنند. اما این مقدار ناچیز از آنتروپی اضافی در حد ترمودینامیکی است، همانطور که می بینید:
${(2V)^{2N}\over (2N)!} = {2^{2N}(N!)^2\over (2N)!}{V^{2N}\over (N!)^2}$به طوری که آنتروپی اضافی از بلند کردن مانع برابر با:ورود به سیستم $\log ({(2N)!\over 2^{2N}(N!)^2})$
شما ممکن است چیزی را در داخل ورود به سیستم تشخیص دهید،${1\over \sqrt{2\pi N}}$
، به طوری که لگاریتم $log(N)$به عنوان ورود به سیستم (n)، آن را گسترده است، و از بین بردن تعداد زیادی.
تغییر آنتروپی در مورد کلی، دقیقا دقیقا توسط لگاریتم نسبت دو حجم فضای پیکربندی قبل و بعد داده می شود:
$e^{\Delta S} = { V_1^N V_2^N \over (N!)^2 } { (2N)! \over (V_1 + V_2)^{2N}} = { V_1^N V_2^N \over ({V_1 + V_2 \over 2})^{2N}} {(2N)!\over 2^{2N}(N!)^2}$
نادیده گرفتن آخرین عامل ترمودینامیکی ناچیز، تغییر ماکروسکوپی در آنتروپی، بخش مناسب به n، این است:
$\Delta S = N\log({4 V_1 V_2 \over (V_1 + V_2)^2})$ همانطور که من محاسبه کردم
ممکن است فکر کنید که عجیب و غریب است که کمی از آنتروپی فقط از این واقعیت بدست آورید که قبل از اینکه دیوار را بلند کنید، می دانستید که عدد ذرات دقیقا N بود، حتی اگر آنتروپی زیر آن است. آیا این بدان معنی نیست که وقتی دیواره را پایین می آورید، آنتروپی را کاهش می دهید، با جلوگیری از مخلوط کردن نیمه راست و نیمه دوم، آنتروپی را کاهش می دهید؟ حتی اگر کاهش آنتروپی کوچک باشد، هنوز قانون دوم را نقض می کند.
هیچ کاهش آنتروپی وجود ندارد، زیرا زمانی که مانع را پایین می آورید، نمی دانید چند مولکول در سمت چپ قرار دارند و چند نفر در سمت راست هستند. اگر شما آنتروپی که نمیدانید به آنتروپی سیستم دیوار پایین را اضافه کنید، دقیقا از دست دادن آنتروپی زیر جلدی حذف می شود. اگر شما سعی می کنید بدانید که چند مولکول در سمت راست قرار دارند، تعداد زیادی از آنها در سمت چپ قرار دارند، شما آنتروپی بیشتری را در فرآیند یادگیری پاسخ به دست می آورید تا از حل ان بدست آورید.
روش دوم
آنتروپی تغییر در مخلوط دو گاز
، اگر دو گازها در ابتدا از هم جدا شوند، در یک جعبه سفت و سخت مخلوط می شوند. من از زیر استفاده می کنم
$\Delta S_1= n_1 c_{v,1} \mathrm{ln}( \frac{T_f}{T_{i,1}}) + n_1 R \mathrm{ln}(\frac{V_1+V_2}{V_1})\tag{A}$
$\Delta S_2= n_2 c_{v,2} \mathrm{ln}( \frac{T_f}{T_{i,2}}) + n_2 R \mathrm{ln}(\frac{V_1+V_2}{V_2})\tag{B}$
و $\Delta S=\Delta S_1+\Delta S_2$
اما من در کتاب درسی که می توانم از فرمول ها استفاده کنم (a)
و (ب)فقط اگر دو گازها متفاوت باشند.
در مورد جایی که من ترکیبی از همان گاز باید استفاده کنم
$\Delta S_1= n_1 c_{v,1} \mathrm{ln}( \frac{T_f}{T_{i,1}}) - n_1 R \mathrm{ln}(\frac{p_f}{p_{i,1}})\tag{C}$
$\Delta S_2= n_2 c_{v,2} \mathrm{ln}( \frac{T_f}{T_{i,2}}) - n_2 R \mathrm{ln}(\frac{p_f}{p_{i,2}})\tag{D}$
در غیر این صورت تغییر در آنتروپی صفر خواهد بود.
از آنجایی که مثال (A) و (C) باید معادل باشند زیرا S یک تابع حالت است، من نمیدانم چگونه میتوان در آن فرمولها برای محاسبه S و دلیل اینکه دو تا از آنها نتیجه درست میدهند و بقیه نتیجه میدهند تفاوتی وجود داشته باشد. نه بازم جواب من این هست آنتروپی تابعی از تعدد حالت است. اگر گاز یکسانی داشته باشید، کثرت حالت را تغییر نمی دهید و بنابراین نمی توانید آنتروپی آن را تغییر دهید. اگر دو گاز متفاوت باشند، فرق می کند
روش سوم
پارادوکس گیبس زمانی بوجود می آید که این دو گاز یکسان باشند. ... اگر دو گاز یکسان در دو دما و فشار یکسان در دو محفظه داشته باشید ، با جدا شدن پارتیشن چیزی تغییر نمی کند - بنابراین نباید تغییری در آنتروپی ایجاد شود.این پارادوکس گیبس است. تناقض با این فرض که ذرات گاز در واقع غیر قابل تشخیص هستند ، حل می شود. این بدان معناست که تمام حالاتی که فقط با یک تغییر ذرات متفاوت هستند باید همان حالت در نظر گرفته شوند.مشتقات آنتروپی به ترتیب با توجه به تعداد ذره و انرژی. عبارت. به عنوان عامل گیبس شناخته می شود. برای محاسبه احتمال ، به یک عادی سازی نیاز داریم.
عامل.$G=H-TS_\text{sys}$ را تعریف کنید. سپس،
$\begin{align}
\mathrm dG &=\mathrm dH-S_\text{sys}\,\mathrm dT-T\,\mathrm dS_\text{sys}\\[3pt]
&=-T\left(\frac{-\mathrm dH}T+\mathrm dS_\text{sys}+S_\text{sys}\,\frac{\mathrm dT}T\right)
\end{align}$در شرایط دما و فشار ثابت ،$\frac{-\mathrm dH}T=\mathrm dS_\text{surr}$ و dT = 0. بنابراین ، ما در پایان با:
$\mathrm dG=-T(\mathrm dS_\text{sys}+\mathrm dS_\text{surr})=-T(\mathrm dS_\text{universe})\tag{roham-01}$با استفاده از $H=U+pV$ و با استفاده از قانون اول ترمودینامیک می توان نشان داد که:$\mathrm dG=V\,\mathrm dp-S\,\mathrm dT$
در ثابت p ، T ، این معادله به dG = 0 کاهش می یابد. با استفاده از این در roham-01 در ثابت T و $\mathrm dG=\mathrm dS_\text{universe}=0$ بازده می دهد.
بنابراین دقیقاً چگونه می توان اصطلاحاً "فرایندهای خود به خودی" را که دارای مقدار منفی dG در ثابت T و p هستند ، بدست آورد؟
تا حدودی می توانم ببینم که چرا نتیجه معقول است: شرایط فرض شده در مشتق شبیه فرایند ترمودینامیکی برگشت پذیر است که برای آن مشخص است $\mathrm dS_\text{universe}$ صفر است.سوال پررنگ اما هنوز بی پاسخ است. شاید من در مورد معنای واقعی ΔG گیج شده باشم.ماهیت آنچه در جریان است این است:معادله شما ، $dG = Vdp -SdT$ ، درست است ، اما محدودیت هایی دارد که شما متوجه آن نمی شوید. به طور خاص ، این فقط برای سیستم هایی اعمال می شود که (الف) بسته هستند (بنابراین هیچ ماده اضافه یا تفریق وجود ندارد) ، (ب) فقط یک جز component دارند (بنابراین بدون اختلاط ، تغییر فاز یا واکنش های شیمیایی) و (ج) که می تواند انجام دهد pV - فقط کار
[* این مورد همچنین برای سیستم های چند جزئی که در آنها ترکیب ثابت است اعمال می شود.]
برای درک اینکه چرا ، در چنین شرایطی ، dT = 0 و $dT = 0 \text{ and } dp = 0 \Rightarrow dG = 0$، بیایید قانون مرحله گیبس را برای چنین سیستمی اعمال کنیم. قانون فاز می گوید:$F = C − P + 2,$
که در آن C تعداد اجزا است ، P تعداد فازهای تعادل است (با p ، فشار اشتباه گرفته نمی شود) ، و F تعداد درجات آزادی است.
از آنجا که C = 1 و P = 1 ، F = 2 بدست می آوریم. این بدان معناست که ما فقط دو درجه آزادی داریم ، یعنی دو روش مستقل که می توانیم مناسبات شدید سیستم را تغییر دهیم. اگر تنها نوع کاری که می توانیم انجام دهیم کار pV است ، تنها راههایی که می توانیم خصوصیات شدید سیستم را تنظیم کنیم تغییر دما یا فشار آن است.
از این رو ، اگر محدودیت های dT = 0 و dp = 0 را روی یک سیستم تک جزئی بسته که فقط اجازه کار pV را می دهیم اعمال کنیم ، سیستم نمی تواند تغییر کند! و اگر سیستم نمی تواند تغییر کند ، مطمئناً $\boldsymbol{dG = 0}$
اما ممکن است اعتراض کنید ، dG حتی در T و p ثابت نیز به طور کلی صفر نیست. بنابراین چگونه این را با آنچه در بالا نوشتیم سازگار کنیم؟ خوب ، ما به عبارتی کلی تر برای dG نیاز داریم که اجازه کار غیر pV ، جمع و تفریق مواد و تغییر در ترکیب را بدهد:$dU = \text{đ}q + \text{đ}w +\sum_i \mu_i dn_i
= \text{đ}q + \text{đ}w(pV) + \text{đ}w (non\text{-}pV) + \sum_i \mu_i dn_i$
از آنجا که می توانیم dU را با استفاده از هر مسیری محاسبه کنیم ، بیایید از یک مسیر برگشت پذیر استفاده کنیم:
$dU = TdS - pdV + \text{đ}w (non\text{-}pV, rev) + \sum_i \mu_i dn_i$
و از:$G = U+pV-TS \Rightarrow dG = dU + pdV +Vdp - TdS - SdT$
$\Rightarrow dG = VdP -SdT+ \text{đ}w (non\text{-}pV, rev) + \sum_i \mu_i dn_i$
در اینجا ،$\sum_i \mu_i dn_i$ مجموع پتانسیل شیمیایی هر گونه$(\mu_i)$برابر تغییر در مقدار گونه $(dn_i)$ است. این تغییر در U و در نتیجه در G است ، زیرا ما ترکیب را تغییر می دهیم
از این رو ، حتی اگر dT = 0 و dp = 0 باشد ، اگر کار غیر pV نداشته باشیم و یا تغییر در ترکیب داشته باشیم (به عنوان مثال مخلوط کردن ، تغییر فاز ، افزایش یا از دست دادن ماده یا واکنش شیمیایی) ، اینگونه نخواهد بود که dG به صفر محدود شود.برای سیستم های دارای ترکیب متغیر (مخلوط)
روش دوم
$\mathrm dU=-p\mathrm dV + dw_{other}+ T\mathrm dS + \sum_i\mu_i\mathrm dn_i$
این منجر به$\mathrm dG = V\mathrm dP -S\mathrm dT + dw_{other}+ \sum_i\mu_i\mathrm dn_i$
یا ، در دما و فشار ثابت ،$\mathrm dG = dw_{other}+ \sum_i\mu_i\mathrm dn_i$
با توجه به مسئله خودانگیختگی ، . برای فرایندی که شامل یک ماده خالص در یک سیستم بسته است و فقط تحت کار انبساط در T و p ثابت است ،$dH=dq$
ولی$dS=\frac{dq_{rev}}{T}$ برچسب "rev" مهم است ، معادله dS = dqT فقط برای هیچ فرایندی برقرار نیست.در T و p ثابت اما بدون محدودیت در برگشت پذیری ،$dS=\frac{dq}{T}$
قانون دوم$dS_{universe} \ge 0$حاکی از آن است
$\begin{align} 0 &\ge -dS_{surroundings}-dS_{system}\\ 0 &\ge \frac{dq}{T} -\frac{dq_{rev}}{T}\\ 0 &\ge dq - dq_{rev} \end{align}$ نتیجه می شود که$dG\le 0$
و فقط برای یک فرایند برگشت پذیر dG = 0 است
